 Introduction
Introduction
RNAScan represents an integrated framework for RNA sequencing (RNA-seq), combining experimental protocols, computational pipelines, and quality control metrics to decode transcriptomes with unprecedented accuracy. Unlike conventional RNA-seq, RNAScan emphasizes end-to-end standardization—from sample preparation to functional annotation—ensuring reproducibility across biomedical, agricultural, and synthetic biology applications. This article details RNAScan’s core workflow, highlighting critical innovations in library construction, sequencing, and bioinformatics analysis.
1. Experimental Design & Sample Preparation
A. Strategic Planning
- Objective Definition: Align RNAScan workflow with study goals (e.g., differential expression, isoform detection, fusion gene identification) (#).
- Biological Replicates: ≥3 replicates per condition to mitigate batch effects and ensure statistical power (#).
- RNA Integrity: Assess RNA quality via RIN (RNA Integrity Number) >8.0 using Agilent Bioanalyzer; degraded samples increase false-negative rates (#).
Suggested Figure: Flowchart: Hypothesis-driven experimental design → Sample randomization → RNA extraction/QC.
2. RNA Library Construction
A. Fragmentation and Reverse Transcription
- RNA Fragmentation: Fragment RNA enzymatically (Mg²⁺/heat) or enzymatically to 200–500 bp (#).
- cDNA Synthesis: Reverse transcribe fragmented RNA using StrandBrite™ kits for strand-specificity (#).
B. Adapter Ligation and Size Selection
- Adapter Design: Y-shaped adapters with Unique Molecular Identifiers (UMIs) to correct PCR biases (#).
- Size Selection: Gel electrophoresis or bead-based purification (e.g., SPRIselect) to exclude fragments <150 bp (#).

Suggested Figure: Library prep workflow: RNA fragmentation → cDNA synthesis → UMI-adapter ligation → Size selection.
3. High-Throughput Sequencing
A. Platform Selection
- Short-Read Platforms (Illumina NovaSeq): 150-bp paired-end reads for expression quantification (#).

- Long-Read Platforms (PacBio Sequel): Full-length isoform resolution (#).
B. Sequencing Depth Optimization
| Application | Recommended Depth |
|---|---|
| Differential Expression | 20–30 million reads/sample |
| Rare Isoform Detection | 50+ million reads/sample |
| De Novo Assembly | 100+ million reads/sample |
4. Bioinformatics Analysis Pipeline
A. Preprocessing and QC
- Adapter Trimming: Tools:
Cutadapt,Trimmomatic(#). - Quality Control: FastQC reports + removal of:
- Low-quality reads (Q < 20)
- Ribosomal RNA contaminants (via
SortMeRNA) (#).
B. Alignment and Quantification
- Alignment Tools:
- Genome Alignment: STAR or HISAT2 for splice-aware mapping (#).
- Transcriptome Alignment: Kallisto/Salmon for rapid pseudoalignment (#).
- Genome Alignment: STAR or HISAT2 for splice-aware mapping (#).
- Quantification: FeatureCounts or RSEM to generate raw count matrices (#).

Suggested Figure: Bioinformatics flowchart: Raw FASTQ → Trimming → Alignment → Count matrix → Downstream analysis.
C. Differential Expression & Functional Annotation
- Statistical Analysis: DESeq2 or edgeR for differential expression (FDR < 0.05) (#).
- Functional Enrichment: g:Profiler for GO/KEGG pathway analysis (#).
5. Unique Advantages of RNAScan
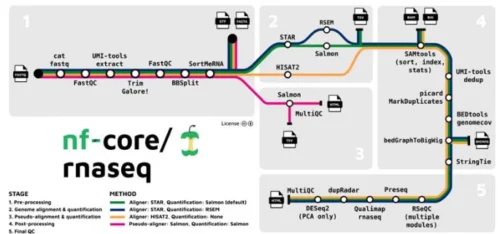
A. Automation via nf-core/rnaseq
End-to-end pipeline integrating:
- Modular Tools: FastQC, STAR, Salmon, MultiQC (#).
- Cloud Compatibility: Executable on AWS/Azure for scalable analysis (#).
B. UMI Integration
- Error Correction: UMIs eliminate PCR duplicates, improving fusion detection accuracy (e.g., KMT2A-PTD in leukemia) (#).
6. Quality Control Checkpoints
| Stage | QC Metric | Tool |
|---|---|---|
| RNA Extraction | RIN > 8.0, 28S/18S ratio > 1.8 | Agilent Bioanalyzer |
| Library Prep | Fragment size distribution | TapeStation |
| Sequencing | Q30 > 85% | FastQC |
| Alignment | Mapping rate > 85% | Qualimap |
7. Applications Across Domains
- Cancer Diagnostics: Detect NTRK fusions with 99% sensitivity using UMI-enhanced RNAScan (#).
- Plant Science: Identify drought-responsive isoforms in Oryza sativa (#).
- Single-Cell RNAScan: Emerging protocols for rare cell-type characterization.
Conclusion
RNAScan redefines RNA-seq by standardizing workflows from sample to insight. Its integration of UMIs, strand-specific libraries, and automated bioinformatics ensures unparalleled accuracy in quantifying transcripts, isoforms, and fusions. As long-read sequencing and AI-based annotation mature, RNAScan will unlock deeper layers of transcriptome complexity—powering precision medicine and engineered biology.
Data Source: Publicly available references.
Contact: chuanchuan810@gmail.com



